

近日,来自荷兰的一个研究团队,在国际顶级学术期刊《自然》上发表了22种转移性实体瘤的全基因组测序研究成果[1]。
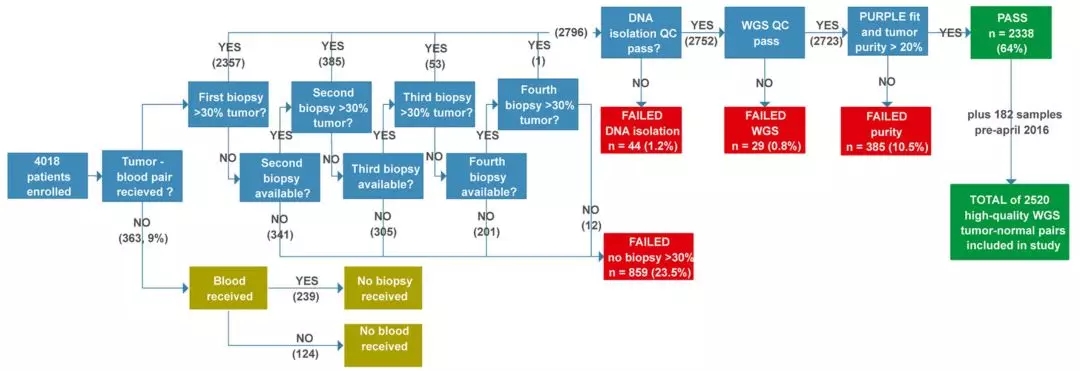
这项由Hartwig医学基金会EdwinCuppen教授领导的研究,对2399位癌症患者的2520对肿瘤和血液样本进行了全基因组测序和特征分析。
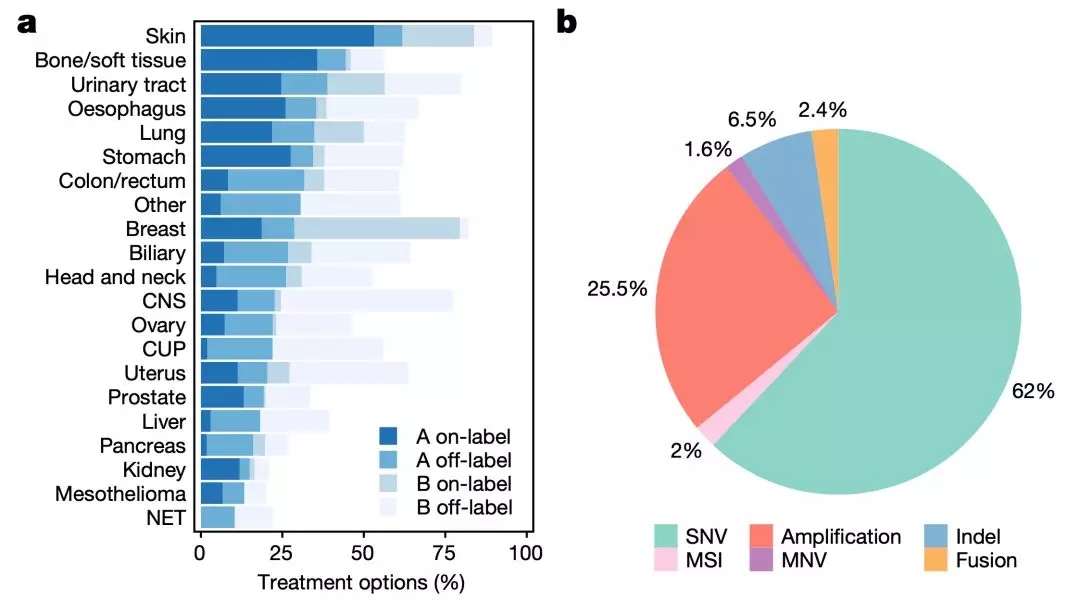
共鉴定出7000多万个体细胞变异,包括近6000万个单核苷酸变异(SNV),84万个多核苷酸变异(MNV),960万个DNA片段插入和缺失(indels),以及65万多个结构变体(SV),并且获得了每个转移肿瘤样本的遗传突变名录。
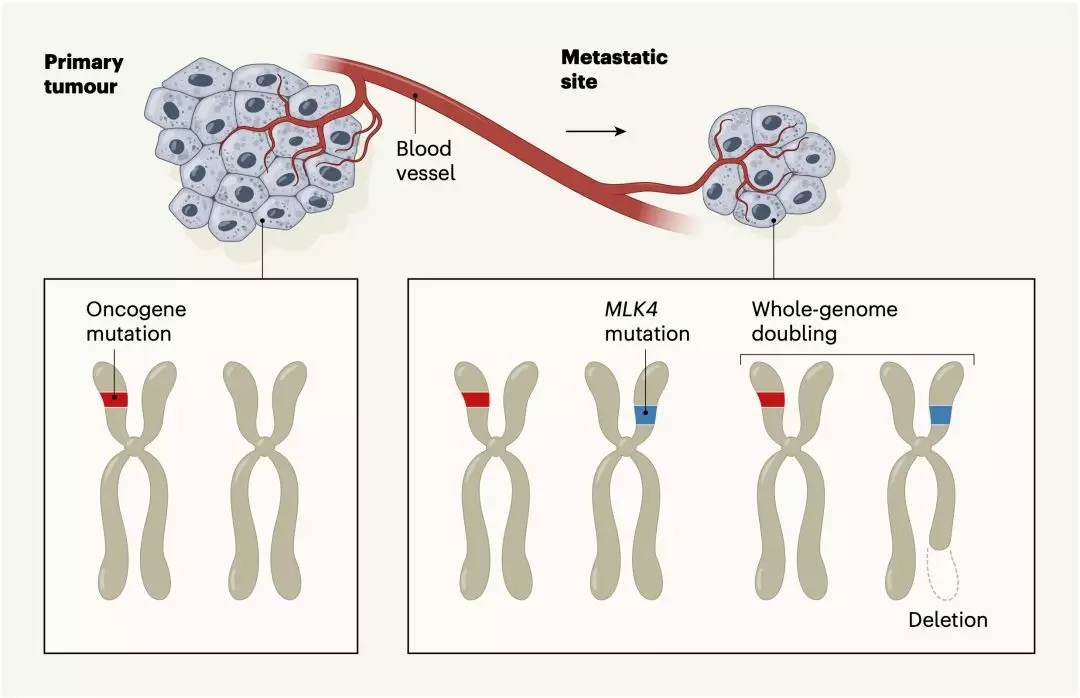
基于上述数据,研究人员发现,可能真的没有驱动癌症转移的特异性突变存在。而且,转移的肿瘤组织中,普遍存在全基因组加倍(WGD)事件,在一些癌种中甚至高达80%,而在原发灶中只有30%。这可能与转移灶对化疗的耐受性相关。
除此之外,研究人员还发现,在单一的转移灶中,肿瘤细胞的异质性非常低,高达96%的基因突变是克隆性的。还有高达80%的抑癌基因通过不同的方式双等位失活。
更为重要的是,研究人员还发现,通过全基因组测序,能帮助62%的患者找到已经获批或者正在开展研究的抗癌药物。
据了解,这也是该领域迄今为止最大的研究。
▲ 探索转移肿瘤的秘密(https://www.nature.com/articles/d41586-019-03123-0)
近年来,测序技术的进步和测序成本的降低,推动了全基因组测序技术在癌症研究中的应用。成年人[2,3]和儿童[4,5]肿瘤的大规模研究,让科学家从分子水平上,对这些癌症有了深入的认知。
这也就顺理成章地推动了基于基因变异开展的癌症治疗[6]。在我们精心打磨的音频课程《医学趋势50讲》中,我们全面地介绍了基因测序在癌症诊疗中的应用,欢迎大家扫描文末二维码试听订阅。
不过,以上的研究大多是在原发肿瘤中开展的,而对于造成90%的癌症死亡的转移,类似的研究却极少。即使有少量转移癌症的研究,要么仅限于特定的癌种[7-9],要么只研究了一些特定的基因[10],要么是外显子层面的研究[11]。
此外,由于随着癌症的进展,原发灶和转移灶都在不停地进化[12,13],也只有全基因组研究能一窥癌症转移的秘密了。因此,对癌症的转移展开全基因组、泛癌种的研究非常有必要。
在荷兰Hartwig医学基金会和个性化癌症治疗中心(CPCT)的统一协调下,荷兰的49家医院参与了这项大规模的研究。截止目前为止,数据库已经累计获取了超4000名患者的数据,还在持续增长中。

▲ 论文非常火爆
接下来,我们就一起来看看研究人员从参与本研究的2399位癌症患者的肿瘤组织中获得了哪些数据。
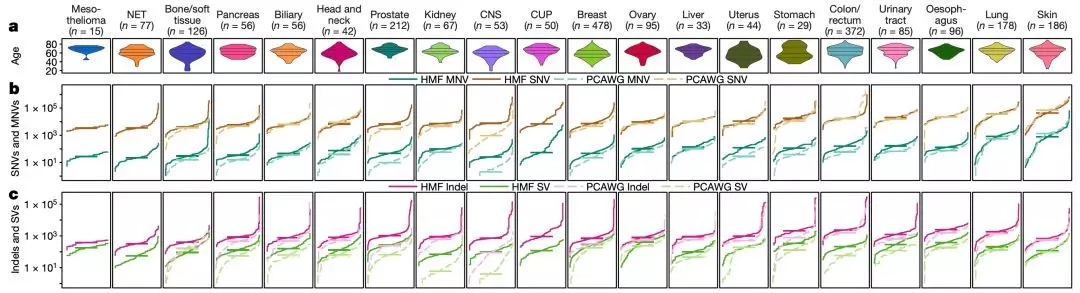
首先是每种癌症的不同类型变异的负担情况。
单核苷酸变异(SNV)的情况:黑色素瘤的中位SNV是44000个,肺癌的是36000,肉瘤的是4100,神经内分泌肿瘤的是3500。这些突变特征,与之前的研究相比,基本匹配[14]。
多核苷酸变异(MNV)的情况:肺癌的中位MNV是821,皮肤癌的是764,这两种癌症的MNV是其他癌种的5倍。这可能与紫外线照射(CC>TT)和吸烟(CC>AA)突变特征比较丰富有关。
插入和缺失(indels)的中值只有SNV的十分之一左右,皮肤癌和肺癌发生率相对较低。
微卫星不稳定(MSI)的发生率:中枢神经肿瘤9.4%,子宫肿瘤9.1%,前列腺肿瘤6.1%。结直肠癌转移灶的MSI仅为4%,低于之前报道的原发性结直肠癌出现频率[15]。
所有肿瘤的中位结构变异(SV)数为193。相对较高的是卵巢肿瘤的412,食管癌的372;较低的是肾癌的71,神经内分泌肿瘤的56。在所有的结构变异中,简单缺失是最常见的,占所有结构变异的33%。除开胃癌和食管癌,缺失性结构性变异在其他肿瘤中比较普遍。
▲ 转移癌症的突变图谱(其实是“天书”)
以上这些结果是转移癌组织与血细胞基因组对比的结果。反映的是转移组织自身的变化,但是没有反映出转移肿瘤组织与原发肿瘤组织之间的差异。
为了了解原发灶和转移灶之间的总体基因组差异,研究人员将他们的这个数据,与迄今最大的原位肿瘤测序数据库PCAWG[16]做了个比较。你没看错,本研究没有分析患者的原发肿瘤基因组数据,对比的数据是其他数据库的数据。
PCAWG这个全基因组测序肿瘤队列,包括2583个肿瘤样本,其中95%的样本是未经治疗的原发肿瘤。
将本研究的数据与PCAWG的数据相比,研究人员发现:单核苷酸变异没有显著差异,意味着单核苷酸变异突变负荷似乎与疾病进展无关,但前列腺癌、乳腺癌和中枢神经肿瘤是例外。
相比之下,转移灶的插入和缺失、多核苷酸变异和结构变异的突变负荷更高,尤其是前列腺癌,研究人员观察到多核苷酸变异,插入和缺失和结构变异的发生率增加了四倍以上。
▲ 患者选择过程
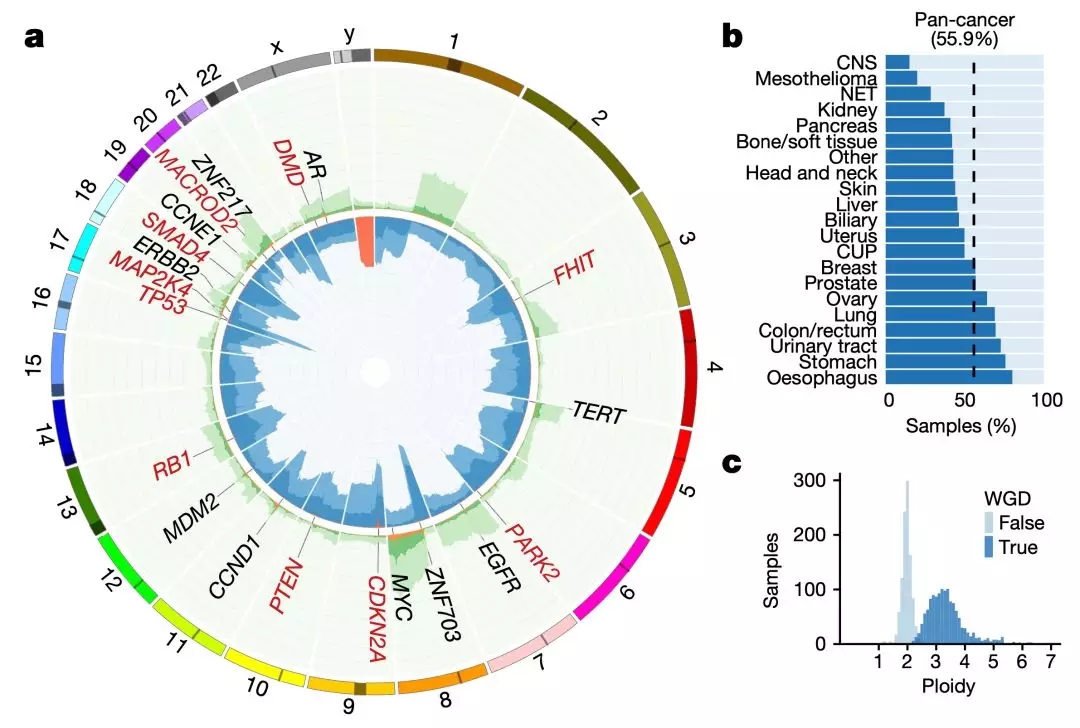
再回过头分析转移肿瘤组织基因拷贝数的变化,研究人员又发现,包含EGFR、CCNE1、CCND1和MDM2这些癌基因的区域,经常发生扩增。另外一个现象是常染色体DNA存在杂合性丢失(LOH),以TP53的LOH出现频率最高,出现在67%的肿瘤样品中,其他的很多抑癌基因也出现LOH。
但没有发现大片段常染色体的纯合缺失,即使是基因纯合缺失也非常罕见。然而,有一个特殊的情况是,Y染色体缺失,36%的男性肿瘤样本存在Y染色体缺失,不过肿瘤之间差异非常大,从中枢神经肿瘤的5%到胆管肿瘤的68%。
还有一个比较常见现象的是全基因组加倍(WGD),56%的肿瘤样本出现了这种现象,从中枢神经肿瘤的15%到食管癌的80%。而且这种情况,远远高于之前报道的原发肿瘤的25%-37%[17-19]。
全基因组加倍在一定程度上增强了癌细胞对化疗的耐药性。而且这种加倍还可以为癌细胞起到缓冲保护作用,防止基因组不稳定、破坏性突变或者染色体片段丢失,给癌细胞带来的致命打击[20]。
▲ 拷贝数变异图谱
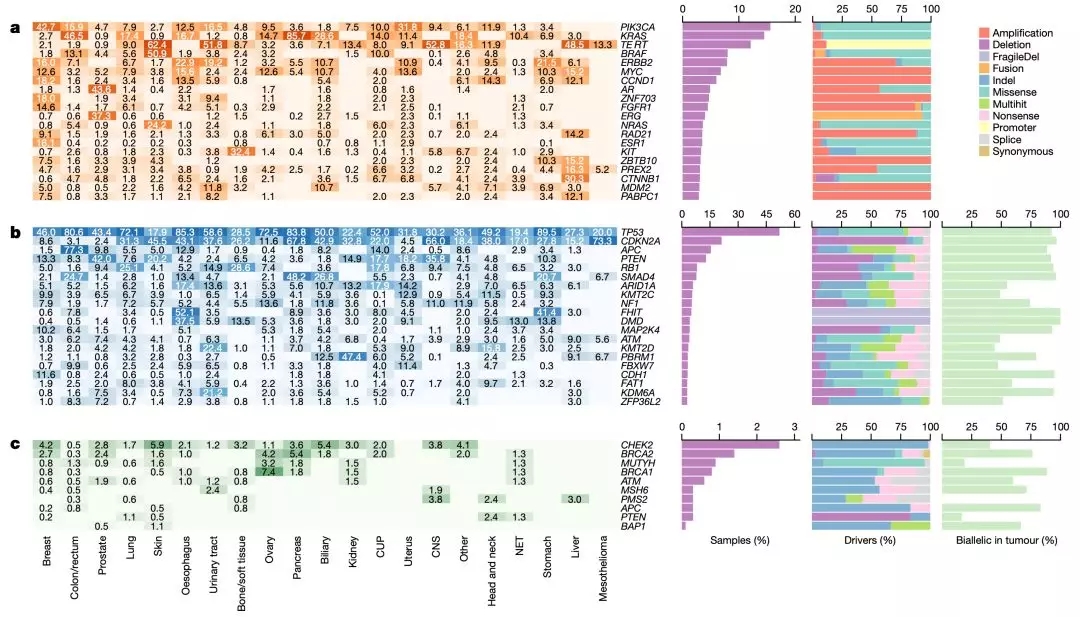
随后,研究人员又给一些关键的癌症驱动突变列了个名录,发现与之前的研究基本相似。其中TP53(52%的样本),CDKN2A(21%),PIK3CA(16%),APC(15%),KRAS(15%),PTEN(13%)和TERT(12%)被确定为最常见的突变基因,它们共同构成目录中所有候选驱动突变的26%。
不过,他们这个名录中排前十的基因突变,在这个队列中的检出率高于原发癌[21]。
遗憾的是,与之前的研究数据库相比较,研究人员只找到了两个有可能驱动癌症转移的基因变异,它俩分别是存在于结直肠癌转移灶样本中的MLK4(又名MAP3K21),和存在于乳腺癌转移灶样本中ZFPM1。这个研究结果再次证实了,“可能不存在转移驱动基因变异”这一猜想。
单从单个患者的角度来看的话,平均每位患者的总驱动突变数量为5.7,其中尿路肿瘤的最高(平均值为8.0),而神经内分泌肿瘤最低(平均值为2.8)。
▲ 常见驱动基因突变
单从抑癌基因的角度看的话,研究人员认为它们的研究结果支持Knudson在1971年提出的双重打击假说[22]。因为80%的抑癌基因驱动突变是双等位失活,也就是说它们彻底完蛋了。这个比例也是迄今为止在癌症全基因组研究中最高的。
对于很多重要的抑癌基因而言,它们的双等位失活率接近100%,例如,TP53(93%),CDKN2A(97%),RB1(94%),PTEN(92%)和SMAD4(96%)。这表明,抑癌基因的双等位失活可能对于癌症的转移非常重要。
研究人员还分析了驱动基因突变成对共存的情况,发现了10对相互排斥的基因组合,以及10对同时存在的基因组合。不过上述的相关性,大部分在之前的研究中已经被建立,只有乳腺癌中有新发现。GATA3和VMP1,以及FOXA1和PIK3CA,总是成对出现;而ESR1和TP53,以及GATA3和TP53,总是有你没我,有我没你。
如果上述关系能在后续的研究中进一步确立,那么就会多出4个治疗乳腺癌的新靶点,而且后面两对似乎是目前大热的协同致死基因对。

▲ 不同癌种驱动变异类型分布
在研究肿瘤的进化动力学过程中,研究人员发现,在整个队列中,只有6.6%的单核苷酸变异(SNV)、多核苷酸变异(MNV)和插入缺失突变,以及3.7%的癌症驱动点突变属于亚克隆。这个比例着实有点儿低。
而且,即使是样本纯度超过80%的样品,亚克隆变异的占比也仅有10.6%。这个比例还是远远低于之前在原发癌中观察到的数据[13]。这就意味着,癌症转移灶非常单一,异质性很低,至少在这个研究中是这样的。
关于这个问题,研究人员也有些其他的思考,可能部分归因于标本的采集方式:因为几乎所有的样本都是通过穿刺活检获得的。如果是因为这个原因的话,液体活检就显得尤为重要了。
基于上述结果,研究人员提出了一个转移模型,他们认为,单个转移灶在任何一个时间点都是由单克隆主导的,有限的肿瘤进化和亚克隆筛选发生在远距离转移之后。当然,他们这个模型不能否认,转移灶中可能存在大量低频的亚克隆。
他们的这个结果与存在多个主要亚克隆的原发肿瘤相反[13,23],不过也与之前的一些认为转移灶中驱动基因异质性低的结果暗合[7,24]。
▲ 临床应用价值
那么发现的这些突变对患者的治疗有没有价值呢?
研究人员发现,在1480名患者(占所有患者的62%)的转移灶肿瘤样本中,至少有一个与用药有关的变异。
其中约一半患者的基因变异,有已经获批或者正在开展临床研究的药物可用,而且没有出现与耐药相关的基因变异。除了上面的50%有药物可用的患者之外,还有31%的患者需要的药物正处于实验室研发阶段。
总的来说,这个大型的队列研究,揭示了一些癌症转移的小秘密,也证明了全基因组测序对癌症精准医疗的重要性。
据悉,这个研究队列的数据是开放的,除了患者的敏感信息之外,所有的数据都可以在数据库的官网调用(https://www.hartwigmedicalfoundation.nl/en/)。
参考资料:
[1].Priestley, P., Baber, J., Lolkema, M.P.et al. Pan-cancer whole-genome analyses of metastatic solid tumours[J]. Nature,2019.
[2].Weinstein J N, Collisson E A, Mills GB, et al. The cancer genome atlas pan-cancer analysis project[J]. NatureGenetics, 2013, 45(10): 1113-1120.
[3].Hudson T J, Anderson W, Aretz A, et al.International network of cancer genome projects[J]. Nature, 2010, 464(7291):993-998.
[4].Grobner S, Worst B C, Weischenfeldt J,et al. The landscape of genomic alterations across childhood cancers[J].Nature, 2018, 555(7696): 321-327.
[5].Ma X, Liu Y, Liu Y, et al. Pan-cancergenome and transcriptome analyses of 1,699 paediatric leukaemias and solidtumours[J]. Nature, 2018, 555(7696): 371-376.
[6].Hyman D M, Taylor B S, Baselga J, etal. Implementing genome-driven oncology[J]. Cell, 2017, 168(4): 584-599.
[7].Yates L R, Knappskog S, Wedge D C, etal. Genomic Evolution of Breast Cancer Metastasis and Relapse[J]. Cancer Cell,2017, 32(2).
[8].Naxerova K, Reiter J G, Brachtel E F,et al. Origins of lymphatic and distant metastases in human colorectalcancer[J]. Science, 2017, 357(6346): 55-60.
[9].Gundem G, Van Loo P, Kremeyer B, et al.The evolutionary history of lethal metastatic prostate cancer[J]. Nature, 2015,520(7547): 353-357.
[10].Zehir A, Benayed R, Shah R, et al.Mutational landscape of metastatic cancer revealed from prospective clinicalsequencing of 10,000 patients[J]. Nature Medicine, 2017, 23(6): 703-713.
[11].Robinson D R, Wu Y M, Lonigro R J, etal. Integrative clinical genomics of metastatic cancer[J]. Nature, 2017,548(7667): 297-303.
[12].Klein C A. Selection and adaptationduring metastatic cancer progression[J]. Nature, 2013, 501(7467): 365-372.
[13].Mcgranahan N, Swanton C. ClonalHeterogeneity and Tumor Evolution: Past, Present, and the Future[J]. Cell,2017, 168(4): 613-628.
[14].Alexandrov L B, Nikzainal S, Wedge DC, et al. Signatures of mutational processes in human cancer[J]. Nature, 2013,500(7463): 415-421.
[15].Gryfe R, Kim H, Hsieh E T, et al.Tumor Microsatellite Instability and Clinical Outcome in Young Patients withColorectal Cancer[J]. The New England Journal of Medicine, 2000, 342(2): 69-77.
[16].Campbell P J, Getz G, Stuart J M, etal. Pan-cancer analysis of whole genomes[J]. bioRxiv, 2017.
[17].Zack T I, Schumacher S E, Carter S L,et al. Pan-cancer patterns of somatic copy number alteration[J]. NatureGenetics, 2013, 45(10): 1134-1140.
[18].Carter S L, Cibulskis K, Helman E, etal. Absolute quantification of somatic DNA alterations in human cancer[J].Nature Biotechnology, 2012, 30(5): 413-421.
[19].Bielski C M, Zehir A, Penson A V, etal. Genome doubling shapes the evolution and prognosis of advanced cancers[J].Nature Genetics, 2018, 50(8): 1189-1195.
[20].Dewhurst S M, McGranahan N, Burrell RA, et al. Tolerance of whole-genome doubling propagates chromosomal instabilityand accelerates cancer genome evolution[J]. Cancer discovery, 2014, 4(2):175-185.
[21].Bailey M H, Tokheim C, Porta-Pardo E,et al. Comprehensive characterization of cancer driver genes and mutations[J].Cell, 2018, 173(2): 371-385. e18.
[22].Knudson A G. Mutation and cancer:statistical study of retinoblastoma[J]. Proceedings of the National Academy ofSciences, 1971, 68(4): 820-823.
[23].Andor N, Graham T A, Jansen M, et al.Pan-cancer analysis of the extent and consequences of intratumorheterogeneity[J]. Nature medicine, 2016, 22(1): 105.
[24].Reiter J G, Makohon-Moore A P, GeroldJ M, et al. Minimal functional driver gene heterogeneity among untreatedmetastases[J]. Science, 2018, 361(6406): 1033-1037.
(来源于: 奇点糕 奇点网)